
El síndrome de QT largo (abreviado SQTL) es una enfermedad cardíaca que puede provocar arritmias malignas y, en algunos casos, muerte súbita cardíaca, especialmente en personas jóvenes y aparentemente sanas.
Es una condición que afecta el sistema eléctrico del corazón y, al igual que el síndrome de Brugada, pertenece al grupo de las canalopatías, trastornos relacionados con los canales iónicos de las células cardíacas. Sobre este ya hablamos en un artículo previo cuya lectura te recomiendo -> ¿Qué es el síndrome de Brugada?
Pero ¿qué es exactamente el síndrome de QT largo?
Un síndrome es un conjunto de signos y síntomas que caracterizan una enfermedad. En este caso, el SQTL se define por una prolongación del intervalo QT en el electrocardiograma (ECG), que refleja un tiempo anormalmente largo en la repolarización de los ventrículos del corazón.
Esta alteración puede desencadenar arritmias peligrosas, como la torsión de puntas. O bien, como dicen los franchutes, «torsades de pointes», una forma de taquicardia ventricular que puede ser fatal. Se llama así en honor a su descubridor François Dessertenne, que lo describió en 1966.
¿Habías oído hablar de este síndrome? Acompáñame a explorar esta condición que, aunque poco común, puede tener consecuencias graves.

¿En qué consiste?
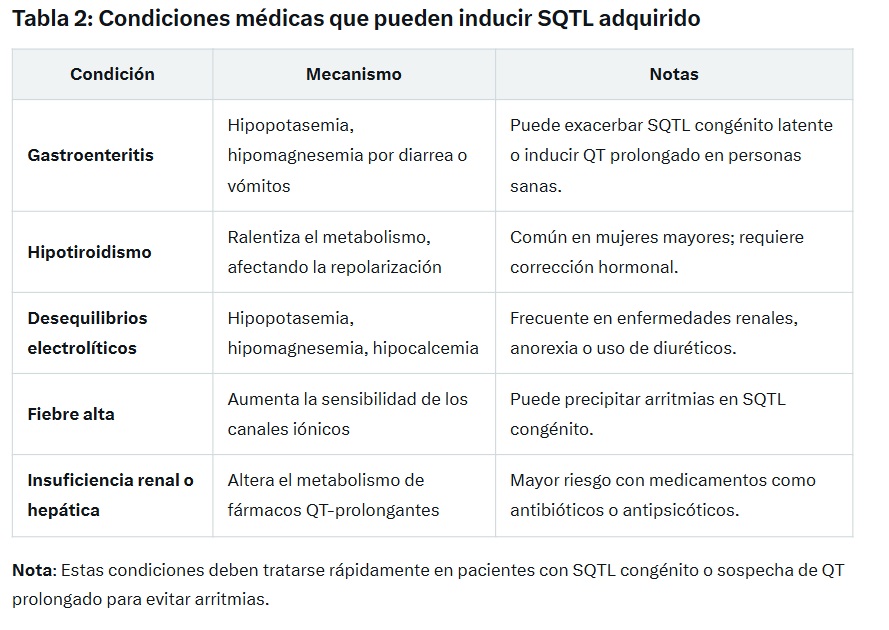
El síndrome de QT largo es una enfermedad genética en la mayoría de los casos, aunque también puede ser adquirido por factores como medicamentos (amiodarona), desequilibrios electrolíticos (iones) o ciertas enfermedades (hipotiroidismo).
A nivel microscópico, afecta los canales iónicos (principalmente de potasio, sodio o calcio) en las células del corazón, lo que altera el equilibrio eléctrico durante la repolarización.
El corazón, a simple vista, puede parecer estructuralmente normal, pero esta alteración en los canales iónicos lo hace eléctricamente inestable, predisponiendo a arritmias que pueden interrumpir el ritmo cardíaco normal.
El corazón se carga y se descarga eléctricamente en cada ciclo cardíaco. A esto se le conoce como despolarización y repolarización. En este caso se afecta la fase de repolarización, es decir, cuando las células miocárdicas recuperan su carga eléctrica. Si esta recuperación de carga se hace lentamente y de forma desigual se pueden provocar arritmias fatales.
El SQTL fue descrito por primera vez en la década de 1950. En 1957, el Dr. Anton Jervell y Fred Lange-Nielsen reportaron un caso de sordera congénita asociada a síncopes y muerte súbita, conocido hoy como síndrome de Jervell y Lange-Nielsen (una forma de SQTL). Más tarde, en los años 60, se identificaron formas no asociadas a sordera, como el síndrome de Romano-Ward, que es la variante más común.

Características del síndrome de QT largo
El SQTL se caracteriza por una prolongación del intervalo QT en el ECG, que mide el tiempo que tarda el corazón en despolarizarse (activarse) y repolarizarse (recuperarse) para el siguiente latido. Este intervalo se mide desde el inicio de la onda Q hasta el final de la onda T.
Cuando el QT corregido (QTc, ajustado por la frecuencia cardíaca) supera los 450-500 ms en hombres o 460-510 ms en mujeres, se considera prolongado. El intervalo QT se usa corregido porque a medida que aumenta la frecuencia cardíaca se acorta. Del mismo modo, cuando la frecuencia cardíaca disminuye se prolonga.
Existen dos tipos principales de QT largo
Congénito:
El congénito se hereda como otras enfermedades genéticas con penetrancia variable. Eso significa que no todos los portadores de la mutación desarrollan síntomas o tienen un QT prolongado en el ECG pero podrían transmitir la enfermedad a la siguiente generación.
Por otra parte, la expresividad variable implica que los síntomas pueden variar ampliamente, incluso dentro de una misma familia. Es decir, que aunque la mutación esté presente y se exprese lo hace dentro de un abanico de mayor a menor intensidad.
- Síndrome de Romano-Ward: Autosómico dominante, más común, sin sordera.
- Síndrome de Jervell y Lange-Nielsen: Autosómico recesivo, asociado a sordera congénita.
- Se han identificado más de 15 genes relacionados, principalmente KCNQ1, KCNH2 (canales de potasio) y SCN5A (canales de sodio). Las mutaciones en estos genes alteran el funcionamiento de los canales iónicos.
Adquirido:
Causado por medicamentos (como antiarrítmicos, antidepresivos, antipsicóticos o antibióticos como eritromicina), hipopotasemia, hipomagnesemia, hipocalcemia, o enfermedades como el hipotiroidismo.

Pueden aparecer arritmias ventriculares por QT largo cuando este intervalo se prolonga por frecuencias cardíacas muy bajas. Normalmente esto es debido a fármacos que prolongan el QT.
Un marcapasos transitorio puede ayudar a acortar el QT si estimulamos el corazón para que lata más rápido. Lógicamente esta estimulación es transitoria y una vez eliminados esos fármacos de la circulación se puede retirar el marcapasos.
¿Es frecuente esta enfermedad?
El SQTL congénito es raro, con una prevalencia estimada de 1 de cada 2.000 a 2.500 personas. Afecta por igual a hombres y mujeres, aunque las mujeres tienen un riesgo ligeramente mayor de eventos arrítmicos, posiblemente por influencias hormonales.
La forma adquirida es más común, especialmente en personas que toman ciertos medicamentos o tienen desequilibrios electrolíticos.
El SQTL puede manifestarse a cualquier edad, pero los síntomas suelen aparecer en la infancia, adolescencia o edad adulta joven (antes de los 40 años). Es una de las principales causas de muerte súbita en jóvenes sin enfermedad cardíaca estructural conocida.
¿Por qué provoca arritmias malignas?
El intervalo QT prolongado indica que los ventrículos tardan más en repolarizarse, lo que crea una «ventana» de vulnerabilidad eléctrica. Durante este período, un estímulo eléctrico prematuro puede desencadenar una torsión de puntas, una taquicardia ventricular polimórfica que puede degenerar en fibrilación ventricular y causar muerte súbita.
Las teorías sobre el mecanismo incluyen:
- Desbalance en los canales iónicos: Las mutaciones en genes como KCNQ1 o KCNH2 reducen la corriente de potasio, prolongando la repolarización.
- Dispersiones regionales: Diferencias en la repolarización entre distintas áreas del corazón pueden generar circuitos eléctricos anómalos.
- En el caso adquirido, los medicamentos o desequilibrios electrolíticos bloquean los canales iónicos, produciendo un efecto similar.
A diferencia del síndrome de Brugada, el SQTL no suele estar asociado con cambios estructurales en el corazón, aunque investigaciones recientes sugieren que algunas formas congénitas podrían tener alteraciones sutiles en el miocardio.
¿Qué síntomas provoca?
Los síntomas del SQTL varían desde la ausencia de síntomas hasta la muerte súbita cardíaca. De ahí que la incertidumbre se apodere de muchos de estos pacientes. Debemos de prestar atención a los siguientes síntomas:
- Síncope (desmayos): Provocado por arritmias como la torsión de puntas, a menudo desencadenado por estrés emocional, ejercicio físico (especialmente natación en el tipo 1), ruidos fuertes (como una alarma en el tipo 2), o durante el sueño/postparto (tipo 3).
- Palpitaciones o sensación de latidos irregulares.
- Mareos o presíncope.
- Muerte súbita cardíaca: Puede ser la primera manifestación, especialmente en casos no diagnosticados.
Aproximadamente el 50% de los pacientes con SQTL congénito son asintomáticos al momento del diagnóstico, detectados por ECG o antecedentes familiares. Factores como la fiebre, el estrés, el ejercicio o ciertos medicamentos pueden precipitar arritmias.

Desencadenantes curiosos del síndrome de QT largo
El SQTL tiene desencadenantes específicos según el subtipo genético:
- SQTL tipo 1 (KCNQ1): La inmersión en agua (como nadar) o el ejercicio intenso pueden inducir arritmias, ya que el estrés físico aumenta la liberación de catecolaminas, alterando los canales de potasio.
- SQTL tipo 2 (KCNH2): Ruidos fuertes (como una alarma o un despertador) pueden provocar un sobresalto que dispara una liberación abrupta de adrenalina, desencadenando arritmias. Esto se debe a la sensibilidad del canal de potasio afectado a estímulos adrenérgicos.
- SQTL tipo 3 (SCN5A): Las arritmias suelen ocurrir en reposo o durante el sueño, posiblemente porque la actividad vagal (parasimpática) predomina, alterando los canales de sodio.
El estrés adrenérgico repentino (por sustos, ejercicio o inmersión en agua fría) activa el sistema nervioso simpático, aumentando la liberación de catecolaminas. Esto puede desestabilizar aún más los canales iónicos defectuosos, creando un entorno propicio para arritmias ventriculares. Por ejemplo, la inmersión en agua fría (como en la natación) combina el estrés físico con un reflejo de inmersión que altera el equilibrio autonómico, un combo peligroso para el SQTL tipo 1.

¿Cómo se diagnostica el síndrome de QT largo?
El diagnóstico se basa en:
Electrocardiograma (ECG):
- Intervalo QTc prolongado (>450 ms en hombres, >460 ms en mujeres).
- Morfología anormal de la onda T (bifida, aplanada o invertida).
- Puede variar en el tiempo, por lo que se recomiendan ECG repetidos.
Pruebas de provocación:
- Prueba de esfuerzo: El ejercicio puede prolongar aún más el QT en algunos subtipos (especialmente tipo 1).
- Prueba de epinefrina: Usada en algunos casos para inducir cambios en el ECG.
Estudio genético:
Identifica mutaciones en genes como KCNQ1, KCNH2 o SCN5A. Útil para confirmar el diagnóstico y guiar el manejo familiar.

Puntuación diagnóstica (criterios de Schwartz):
Combina hallazgos de ECG, antecedentes clínicos (síncope, muerte súbita familiar) y antecedentes familiares para estimar la probabilidad de SQTL.
El diagnóstico es más probable en personas con antecedentes familiares de SQTL, muerte súbita o síncope inexplicado, especialmente si se acompaña de un ECG anormal.
¿Cuál es el tratamiento del síndrome de QT largo?
El tratamiento depende del riesgo de arritmias del paciente, evaluado según síntomas, antecedentes familiares y hallazgos en el ECG o pruebas genéticas.
Medidas generales:
- Evitar desencadenantes: Medicamentos que prolongan el QT (consultar listas en crediblemeds.org), deportes competitivos (especialmente en tipo 1), o situaciones de estrés intenso. No obstante, en cuanto al ejercicio físico se debe individualizar según el caso.
- Corregir desequilibrios en los iones (potasio, magnesio, calcio). También los procesos que pueden estar causando esas alteraciones como por ejemplo problemas gastrointestinales. Ayudar a eliminar los fármacos que prolongan el QT.
- Marcapasos transitorio: en casos en los que por el efecto de un fármaco que deprime la frecuencia cardíaca el corazón está lento y se prolonga el intervalo QT. La estimulación con un marcapasos de manera transitoria puede evitar las arritmias ventriculares hasta que se metabolicen y se vayan eliminando esos fármacos.
Farmacoterapia:
- Betabloqueantes (como propranolol o nadolol): Reducen el riesgo de arritmias en el 70-80% de los casos, especialmente en tipo 1 y 2.
- Mexiletina: Útil en el tipo 3 (mutaciones en SCN5A). Este fármaco es especialmente útil no solamente en este tipo 3 sino también en otros casos en los que la frecuencia cardíaca es muy lenta. Ahí los betabloqueantes la pueden reducir todavía más y existe el riesgo de incrementar el QT por la propia bradicardia.
Desfibrilador implantable (DAI):
- Indicado en pacientes de alto riesgo (síncope recurrente, paro cardíaco previo, o QTc muy prolongado a pesar de betabloqueantes).
- Puede salvar vidas, pero conlleva riesgos como infecciones o descargas inapropiadas.

Denervación simpática cardíaca izquierda:
Lógicamente es un tratamiento de tercer nivel. Primera línea serían las recomendaciones generales, junto con los betabloqueantes. El DAI se reservaría para casos de alto riesgo. Y la denervación simpática es un procedimiento quirúrgico para pacientes con arritmias recurrentes a pesar de haberse agotado líneas previas de tratamiento.
Asesoramiento genético:
- Esencial para identificar portadores en la familia y guiar el manejo.
- Los pacientes asintomáticos con QTc prolongado suelen ser monitoreados, pero pueden requerir betabloqueantes si tienen alto riesgo genético o familiar.
Ejercico físico en el síndrome de QT largo
Recientemente se publicaron los resultados del estudio LIVE-LQTS (Lifestyle and Exercise in the Long QT Syndrome). Se incluyeron individuos con diagnóstico de QT largo y se dividieron en dos grupos.
Eran 1413 participantes de 8 a 60 años y el 52% realizó ejercicio vigoroso, entendido como más de 60 horas al año de ejercicio superior a 6 METS. 1 MET es la energía que gasta tu organismo simplemente por el hecho de existir, o sea, tu gasto energético basal. Una clase de aeróbic, correr en cinta a partir de 8 km/h, ciclismo moderado o caminar cuesta arriba ya son actividades de intensidad moderada a vigorosa.
Pues bien, tras tres años de seguimiento la tasa de eventos cardíacos fue de 2,6% en el grupo de ejercicio vigoroso y del 2,7% en el grupo de ejercicio no vigoroso. Concluyeron que en general, las tasas de eventos en el síndrome de QT largo eran bajas por norma habitual.
Y que no se demostraron más eventos en las personas que realizaban ejercicio vigoroso. Esto no establece una regla general, pero sí para factible la realización de ejercicio físcio de alta intensidad bajo supervisión médica adecuada. Y es importante individualizar las recomendaciones de ejercicio físico para cada paciente.
Pacientes con QT persistentemente prolongado por encima de 500ms, síncopes de repetición, STQL tipo 1, intolerancia a betabloqueantes o baja adherencia a los mismos serán pacientes en los que se siga desaconsejando una actividad física intensa.

En definitiva
El síndrome de QT largo es una condición potencialmente grave, pero con un diagnóstico y manejo adecuados, muchos pacientes pueden llevar una vida normal.
Su prevalencia es baja, pero su impacto puede ser devastador si no se detecta a tiempo. La muerte súbita puede ser la primera manifestación, especialmente en jóvenes, lo que subraya la importancia de investigar síncopes o antecedentes familiares de muerte súbita.
El manejo involucra a cardiólogos, arritmólogos y genetistas. Los betabloqueantes y el DAI son las terapias principales, junto con la prevención de desencadenantes.
Además, es fundamental que los pacientes, sus familias y entorno cercano conozcan las maniobras de reanimación cardiopulmonar (RCP), un conocimiento que debería ser universal.
¿Habías oído hablar del síndrome de QT largo? Si tienes comentarios o quieres compartir experiencias, ¡te leo!